シーケンスタグ30周年
今年はシーケンスタグ検索が誕生してから30年にあたります。この技術は、欧州分子生物学研究所(EMBL)のタンパク質・ペプチドグループに在籍していたMatthias MannとMatthias Wilmによって開発されました。EMBLは創立50周年を迎え、その間、科学の重要な拠点としてあり続けました。シーケンスタグの論文が発表されたのは1994年で、Mann研究室のマイクロエレクトロスプレーの論文と同じ年でした。この2つの技術は同時期に手を携え、ペプチドやタンパク質の同定を可能にする技術となりました。
元の論文には、配列タグを説明する素晴らしい図があります:
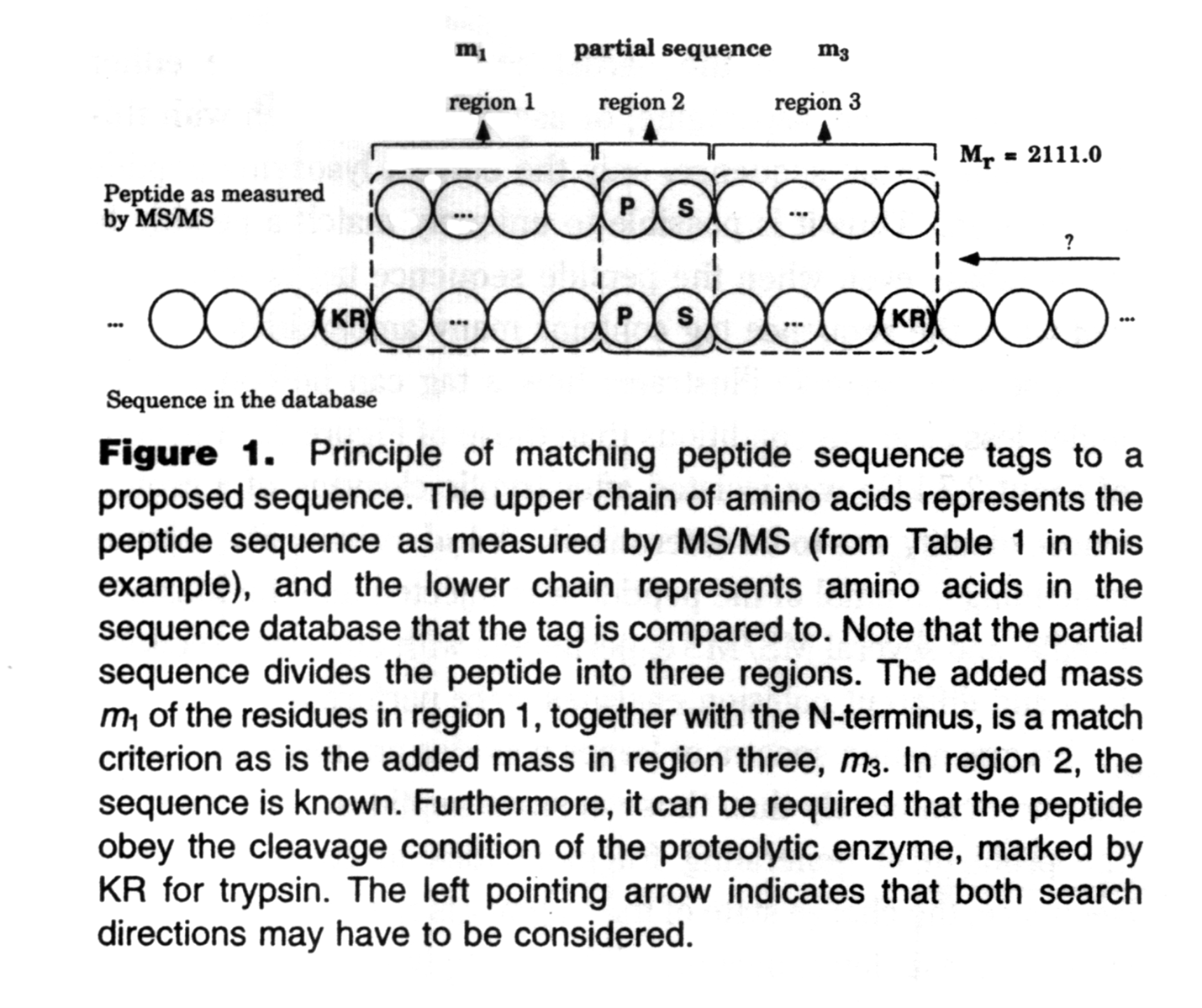
Mascotサーバーでは、リリース最初のバージョンから「Sequence tag」検索、すなわちスペクトル情報と配列情報の混合情報からペプチドを同定する方法、をサポートしていました。昔は簡単にアクセス可能で、配列タグとそれに関連する情報を組み合わせた検索を行う事ができる検索エンジンがいくつもありました。しかし現在もこの機能を提供しているのは私が知る限りProtein ProspectorとMascot Serverだけとなりました。もし配列タグの定義やその処理内容について知りたい場合は、Sequence Queriesのチュートリアルをご覧ください。
私は幸運にも1994年にEMBLのタンパク質・ペプチドグループでペプチドケミストとして働いていたので、当時の開発についてもう少し詳しく説明することができます。消化酵素処理されたタンパク質は、C18カラム材料が充填されたガラス針で脱塩され、Sciex API III質量分析計のエレクトロスプレーソースとして使用される第2の針にスピンされます。マイクロエレクトロスプレー(後にナノエレクトロスプレーと改名)は画期的な進歩をもたらしました。それは、1μLの試料の測定が、当初装置が備えていたエレクトロスプレー源ではせいぜい1~2分であったものを、40分以上と劇的に延ばした事です。MS/MSデータの取得中、測定対象となるペプチドを手動で選択します。十分なMS/MSデータが集まり次第、次のペプチドを選択していきます。サンプルはどんどん消費されていくので、解析は時間との闘いでもありました。今振り返ると非常にエキサイティングなプロセスでした。
当時研究室には配列タグを自動作成するソフトウェアがなかったので、データは手作業で処理されていました。プロセスをスピードアップするため、アップルスクリプトを使ってスペクトルを一定の大きさ(ダルトン)単位のウィンドウに再整理し、1ペプチドにつき2~4ページほど印刷をしました。加えて、アミノ酸の質量が記された定規を準備しました。その定規はOHPでよく使われる透明なアセテートに印刷していました。大きなピークを出発地点として、定規を使ってアミノ酸質量差に一致する次のピークを選び、同じイオン系列である複数のアミノ酸の「はしご」を作っていきます。3つ以上のアミノ酸が揃ったら、それに2つの末端質量を組み合わせて検索用のタグを完成させます。そのタグを、同じくMatthias Mannとその研究室が開発したPeptideSearch配列タグ検索エンジンに入力して検索を行います。研究者側が経験を積むと、スペクトルを測定している間にタグを読み取り、次の標的ペプチドを選択する前に検索をかける、といった作業の効率化も進みました。
配列タグが発表された当時から、上記のようにわざわざ人間の解釈を介さずに自動化された検索方法があることが指摘されていました。LC-MS/MS法が確立されると、Sequence Tag法は時代遅れな存在となりました。
Mascot Distillerを使い、手動でシーケンスタグを作成する実際のプロセスは、昔の方法と非常によく似ています。以下の図はBruker timsTOFのデータです。m/z1279 のピークを選択し、そこから解析によりタグを作成しました:
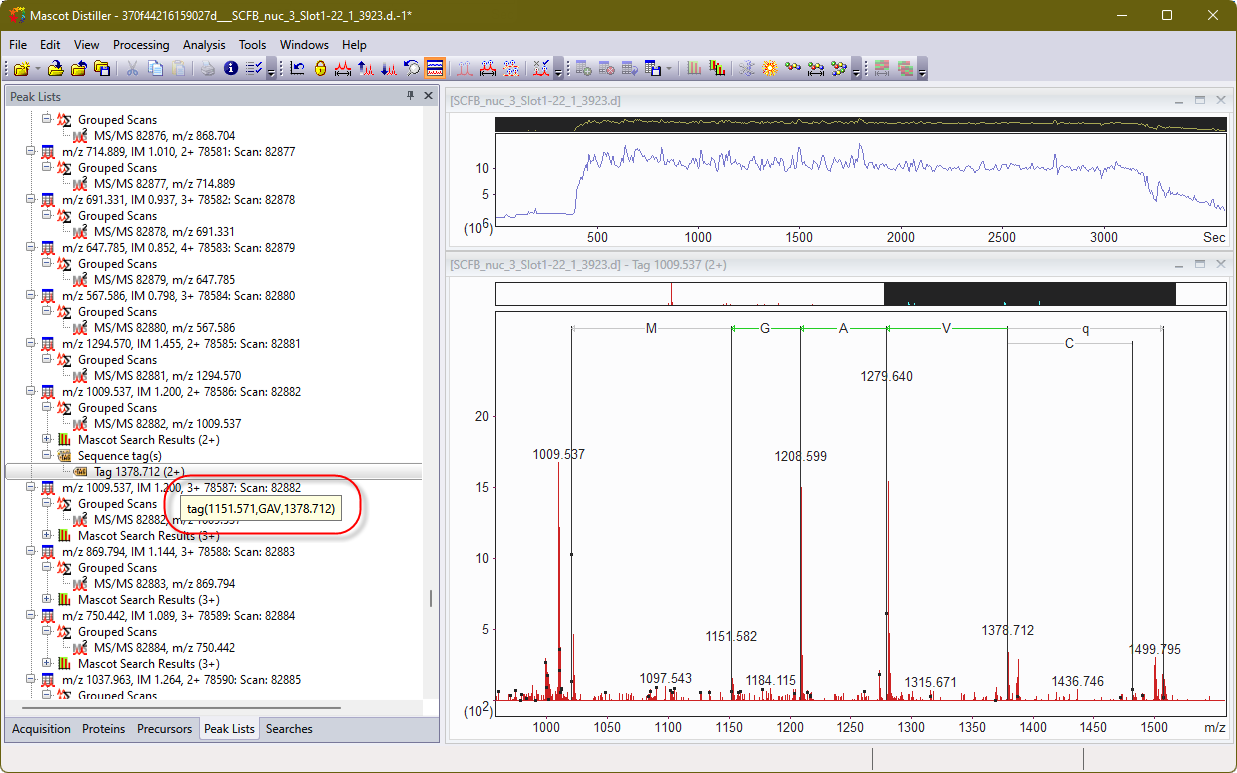
シーケンスタグを定義するのは手作業で困難であるにもかかわらず、なぜMascotサーバーはいまだにこの解析方法をサポートしているのでしょうか?その答えは、弊社がError tolerant tag(etag)検索と呼んでいる検索方法にあります。Error tolerant tag検索は、予期せぬ修飾や一塩基多型など配列のわずかな違いを許容した検索を行う事ができます。etagによる検索が行われた時はペプチド質量の絞り込みを一定レベルで緩和させる事ができます。フラグメントイオンの質量値は上記2つの可能性のいずれかに適合したケースを考慮する事が出来ます。両方の値が変更されないか、両方の値がペプチド質量と同じ量だけシフトされます。
別の選択肢もあります。それは、配列タグを手動で作成するのではなく、Mascot Distiller のsearch toolbox de novo アルゴリズムを使用する方法です。通常のMASCOT検索で有意な一致が得られなかったスペクトルに対してこの方法を適用する事で、答えが見つかる可能性があります。
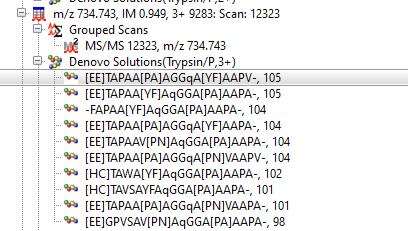
この方法ではde novo計算結果からetagsを準備してMASCOT検索することができます。例えば上の図にあるようなde novo検索結果から生成されたetagの例を以下にご紹介します。
2201.207624 from(2202.214900,1+) title(9283: Scan 12323 (rt=11.8765, p=9379) index(0) etag(1944.16966,TAP,1674.99221) etag(1772.04349,PAA,1532.91628) etag(1293.79272,GG[Q|K],1051.56527)
実際に検索すると、それぞれのクエリーから似たようなスコアの検索結果がいくつも出てきます。その中で、"PAA "配列のタグを使った検索結果は興味深く、説明も簡単そうな内容でしたので紹介します。
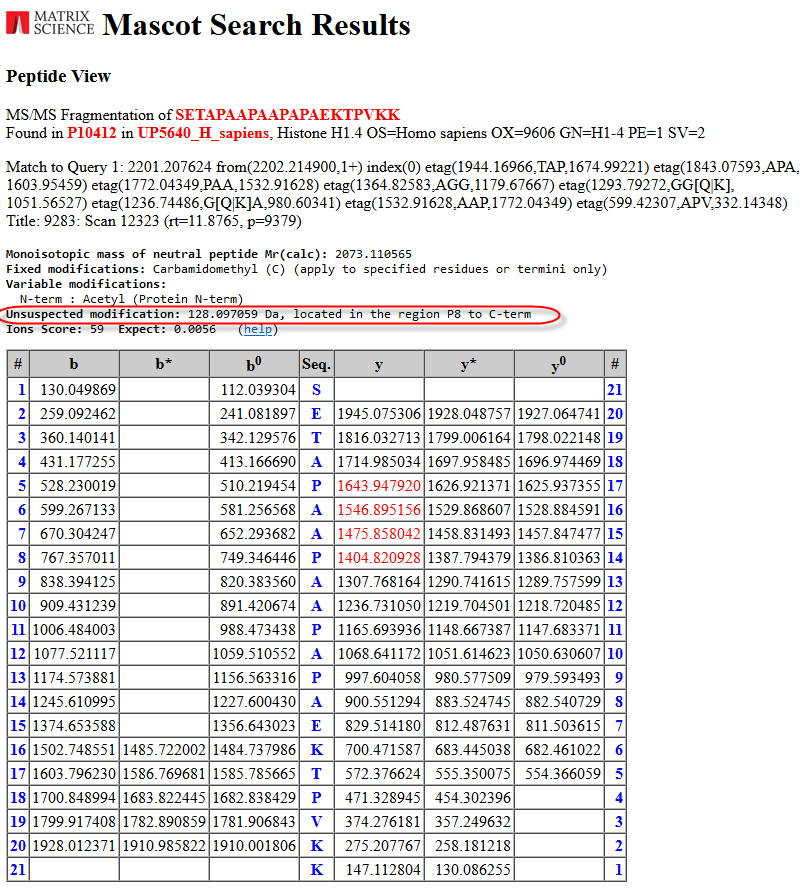
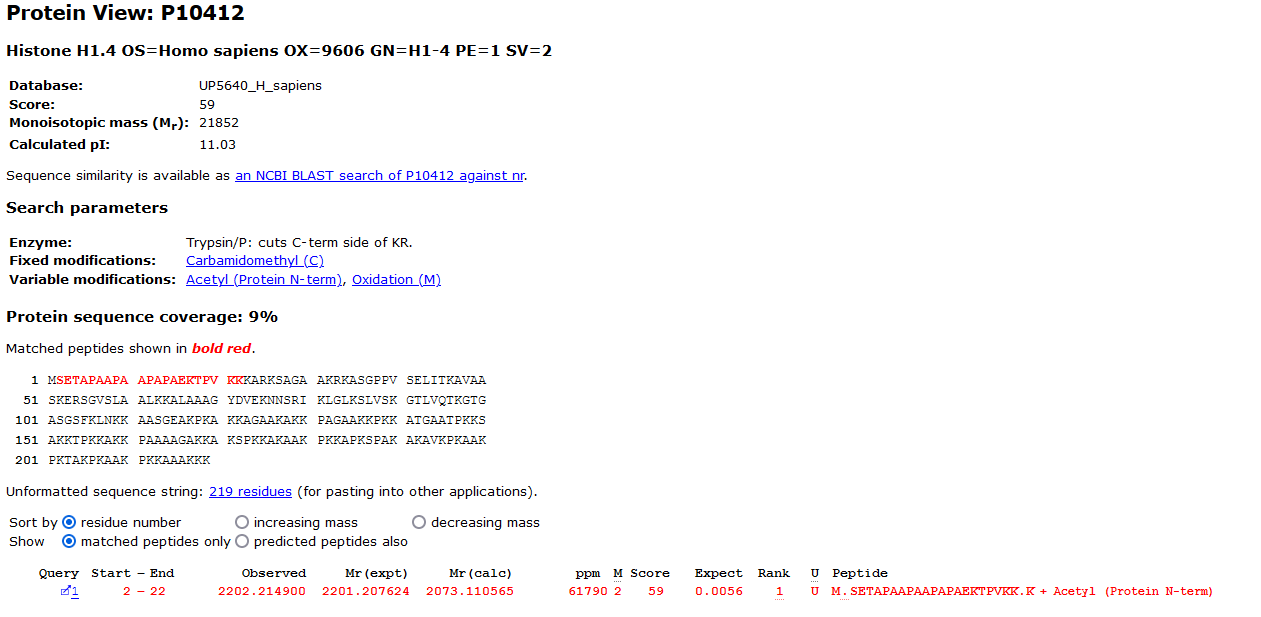
(上の図の赤い丸の部分は)P8からC末端の間に、128.09Da(おそらくリシン)の質量の修飾がついているという可能性を示す結果が表示されています。タグの中の質量はすべてこの修飾の質量分だけシフトします。Protein Viewのレポート(下図)から、タンパク質中におけるこのペプチドの次のアミノ酸は実際にはリシンであることがわかります(訳者注:「本来演算の想定範囲外だったが、C末端にもう1つKまで配列が延びたペプチドを測定した結果である事が示唆されています」という意味です):
この結果を踏まえスペクトルをもう一度チェックしました。最初のMS/MS検索で同定されなかったのは、キメラ(訳者注:複数のペプチドが混在したスペクトル)であり、未同定の大きなピークがたくさんあるからだと考えられます。ピーク抽出の設定を変更し、キメラである事を前提とした再検索を行えば、より簡単に、より多くのペプチドを同定できるかもしれません。
エラートレラントシーケンスタグ検索は、標準検索、エラートレラント検索、de novoシーケンスの後に行うべき、検索戦略の最後の手段といえるでしょう。結果の一部が同定できず問題があると考えた場合、etagsはツールボックスに入れておくと便利なテクニックではないでしょうか。
シーケンスタグが導入された頃、手作業でシーケンス用のペプチドをチェックしていた経験がありますか?もしよろしければコメントにその当時の思い出を投稿してください。
Keywords: anniversary, de novo, error tolerant, history, Mascot Distiller