チュートリアル:最適なDeepLCモデルの選択
Mascot Server 3.0は、予測保持時間の情報を利用してデータベース検索の判定を改善することができます。予測はDeepLCによって実行されます。Mascotでは勾配の長さ、カラムタイプ、ペプチドの特性が異なる20種類のモデルが予め準備されています。このチュートリアルでは、実験に最適なモデルの選択方法について説明します。
DeepLCとは?
DeepLCは、畳み込みニューラルネットワーク(深層学習)アーキテクチャを使用する保持時間予測ツールです。2021年にNature Methods誌で発表された論文には、このアーキテクチャと設計上の選択肢について非常に優れた説明が記載されています。簡単に説明すると、DeepLCは保持時間の予測に原子組成とアミノ酸エンコードを使用しています。そのためトレーニングデータに含まれない修飾を持つペプチドについても正確な予測を行うことができます。
MascotにおけるRT予測の目的は何でしょうか?
「予測保持時間」は、正しいマッチと誤ったマッチを区別するために使用される多くの特徴(feature)の1つです。区別の能力を上げるため、特徴(feature)として実際に利用されるのは予測保持時間と観測された保持時間の差です。保持時間はMS/MSスペクトルから計算された指標と直交しているため、この特徴(feature)は非常に強力な改善をもたらす可能性を秘めています(訳者注:「マススペクトルと保持時間とはその内容的に大きく性質が異なり、互いに影響を及ぼしている要素が非常に少ないので、正解不正解の区別のためのパラメーターとして両方を採用する事には大きな意義がある」、という意味)。適切なDeepLCモデルを選択すると、誤ったマッチの保持時間差の値は広く分散する一方、正しいマッチの保持時間差はゼロの周辺に集まることが期待されます。このようなクラスタリングは、Percolatorの半教師あり学習に最適です。
DeepLC の使用方法
まず、保持時間の情報がMGFファイルの「RTINSECONDS行に含まれていることを確認してください。「title」行の一部に保持時間情報が含まれているだけでは不十分です。 MascotDistiller なら適切な入力データを作成する事ができます。mzML を使用している場合、Mascot は CV(controlled vocabulary)のMS:1000016 および MS:1000826の内容を確認します。
上記の事を簡単に確認するためには、検索結果を開いてください。ペプチドと有意なマッチをしたデータを選択し、Peptide Viewレポートを開いて、一番上にあるrawデータに関する情報を参照してください。「rtinseconds」という行を探し、以下のように記載されているかご確認ください。
Match to Query 244: 741.448728 from(371.731640,2+) intensity(325195.44)
scans(4802) rawscans(sn4802) rtinseconds(2802.2659) index(3027)
このように情報が含まれていれば、実測の保持時間を正しく認識し、DeepLCを使用できる状態です。
Mascot Server 3.0 はMS2Rescoreプログラムと統合されており、DeepLC はそのコンポーネントの1つです。 DeepLC のニューラルネットワークは使用前にトレーニングする必要がありますが、Mascot Server 3.0 には20のトレーニング済みモデルが付属しています。データベース検索後、結果レポートのフォーマットコントロールからモデルを選択・変更してMS2Rescoreを実行する事ができます。

DeepLC モデルの命名規則は?
MascotはDeepLCのアップストリームバージョンと同じモデル名を使用します。命名規則については、DeepLC GitHubページ説明があります。
[full_hc]_[dataset]_[fixed_mods]_[hash].hdf5
Mascotに含まれるすべてのモデルには「full_hc」という接頭辞が付いています。これは単に「学習済みで最適化されたモデル」であることを意味します。 [dataset] はモデルの学習に使用されたデータセットの略称です。「fixed_mods」はフラグで、これが存在する場合、fixedの修飾データが学習データに含まれていた事を示します。 [hash] はモデル実装内容の詳細です。
モデルの命名は少しわかりにくい点もあります。Mascotではモデルの選択に役立つよう、勾配の長さなどのより関連性の高い属性を補足説明した表を準備しています。
良いモデルの選び方は?
まず、あなたの実験データにおいて、クロマトグラフィーが再現可能・一貫性があることを確認してください。再現性がない場合は、いくら機械学習をしてもデータは改善されません。
実験データについて、以下の情報を確認してください。勾配の長さと、種類は逆相(RP)、HILIC、またはその他の方法なのかどうか?isobaric な定量(TMT/iTRAQ)、metabolicな定量(SILAC)、あるいは両方、を使用したか?サンプルはリン酸化のような特定の修飾を多く含むのか?
DeepLCの開発者は、最適なモデルを選ぶ最初のステップとしてfull_hc_PXD005573_mcpを使用する事を推奨しています。このモデルは、ヒトのトリプシン消化ペプチドでカルバミドメチル化(C)と、酸化(M)およびアセチル化(タンパク質N-末端)の解析データでトレーニングされています。カラムの種類は逆相(RP)で、溶出時間は2時間です。これらの実験条件がぴったり一致するのであれば勿論、このモデルはあなたのデータ解析に非常に適合します。
DeepLCはこれらの条件が完全に一致しないようなケースにおいてもかなり寛容です。例えば溶出が2時間より少し短いまたは長い場合、ペプチドが異なる生物種由来の場合、修飾がわずかに異なるがトレーニングデータと化学的に類似している場合など、要因の1つが異なっていても、full_hc_PXD005573_mcpは依然として良い選択となります。ただし、条件がトレーニングデータの内容から離れるほど、予測の精度は低下します。
Mascotには20種類のモデルが搭載されていますが、順番にすべてを試す必要はありません。最適なモデルを選別するために推奨される手順は以下の通りです。
- まず、DeepLCモデルの一覧から、不適切なカラムタイプを持つモデルを除外して候補のリストを短くします。
- 次に、勾配がご自身のものと大きく異なるモデルを除外します。勾配が30分の場合、2時間または4時間のモデルよりも1時間のモデルの方が適しています。逆に、ご自身のデータが長い勾配を使用していた場合、短い勾配でトレーニングされたモデルは避けるべきです。
- 最後に、トレーニングセットで使用されたペプチドの性質を考慮します。サンプルにTMTラベルまたはリン酸化がある場合、それらを含むモデルを最終候補リストに含めます。そしてもし完全に適合するモデルがない場合、モデルを1つに絞り込むことはできず、いくつかのモデルを試す必要があるでしょう。
モデルを選択する上で、2つの重要な評価基準をチェックして選択項目の妥当性を評価してください。それらの値は機械学習の品質レポート画面、’Rescoring features’タブ内にあります。
1. 保持時間について、予測値と観測値の散布図を確認してください。予測値は対角線上に位置していますか? もし逸脱している場合は、どの程度のばらつきがありますか? 勾配の開始時と終了時の予測値もまた、対角線上にありますか?
2. 相対平均絶対誤差(RMAE)を確認します:お客様のデータセットは、DeepLCベンチマークデータセットの選択と比較してどうでしょうか?通常、RMAEは2%以下が目標ですが、3~5%程度であれば許容範囲内です。相対誤差が5%を超える場合、散布図では通常、大量の散布または非線形挙動として視認できます。
実験条件に適したモデルが見つかれば、手順を繰り返す必要はありません。デフォルト設定としてDaemonのパラメータエディタで該当モデルの選択を保存するか、検索フォームのデフォルトとしてクッキーに保存してください。今後のデータベース検索にも同じモデルを使用する事ができます。
誤ったモデルを選択した場合、何が起こるのでしょうか?
あまり適合していないモデルを選択した場合について少し話を掘り下げます。幸いなことに、結果が悪くなることはほとんどありません。例えば、モデルがすべてのペプチドに対して同じ保持時間を予測したとします。その場合、ターゲットとデコイのマッチに対するRTの差は極端にばらつくので、Percolatorはこれらを区別するためにRTの差を使用しないはずです。RTの情報を使っても両者を分離できないからです。
すべてのデータセットに「最適」なモデルは存在しますが、DeepLCモデルのほとんどは、幅広いデータセットに対して許容できると言える程度の結果を生み出します。2021年のDeepLC論文の補足情報の図S-7で示されているように、著者らは、トレーニングに使用したすべてのデータセットに適用したすべてのモデルについて、予測された保持時間と観測された保持時間の相関を計算しました。良好な適合をもたらす最も重要なポイントは「カラムタイプ」です。SCXの保持時間はRPとは大きく異なり、RPはHILICとは異なります。しかし、ほとんどのRPモデルはほとんどのRPデータセットで許容できる程度の改善性能を発揮します。
今回、上記の点について弊社でも改めて解析を行いました。以下の一覧はMascotに同梱されているすべてのモデルを適用した際の、RMAEとRT散布図をまとめたものです。データセットは、8月のブログ記事(英語版、日本語版)でも使用されたCPTAC酵母サンプルの検索です。
配列FDR1%を基準として有意なユニーク配列の数をカウントしています。比較対象とするベースラインは、DeepLCもMS2PIPモデルも選択しないでPercolatorを有効にした検索結果です。有意な配列の数は2740個で、表中の有意なユニークペプチド配列の増加数は、純粋にDeepLCの適用によりもたらされたことになります。
| Model | Sig. unique sequences |
RMAE | Scatterplot | |
|---|---|---|---|---|
| full_hc_unmod_fixed_mods | 2979 (+8.7%) | 3% |
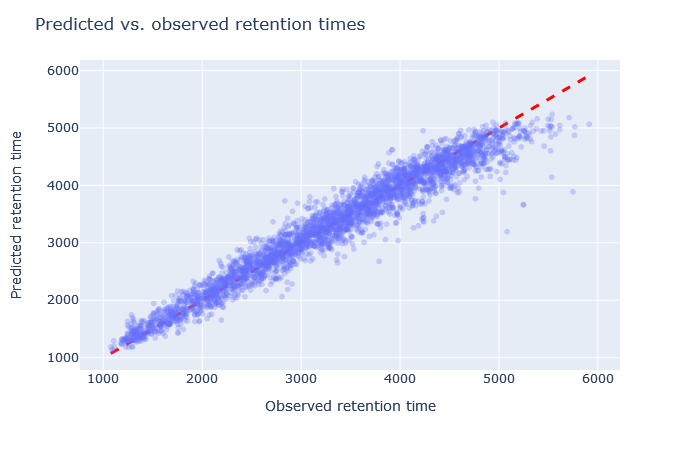
|
(results report) |
| full_hc_dia_fixed_mods | 2971 (+8.4%) | 3% |
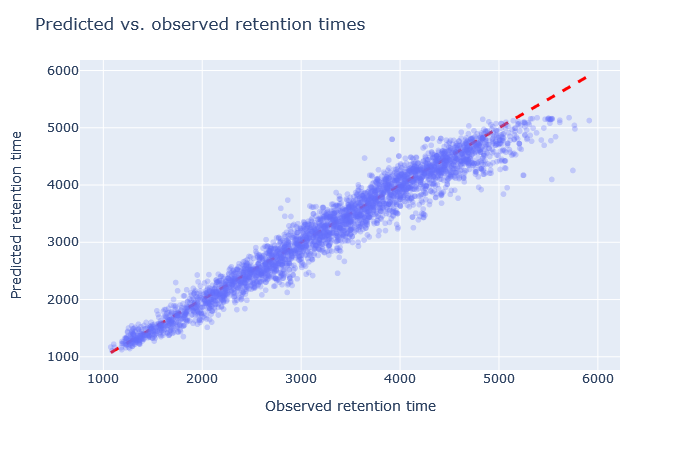
|
(results report) |
| full_hc_hela_lumos_2h_psms_aligned | 2947 (+7.6%) | 2.5% |
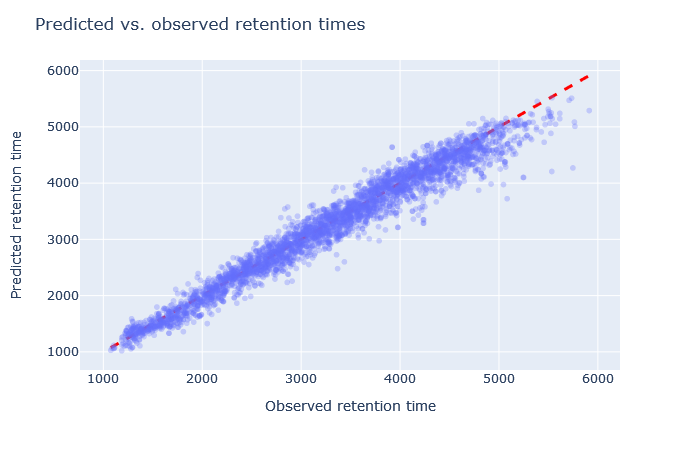
|
(results report) |
| full_hc_hela_hf_psms_aligned | 2943 (+7.4%) | 4% |
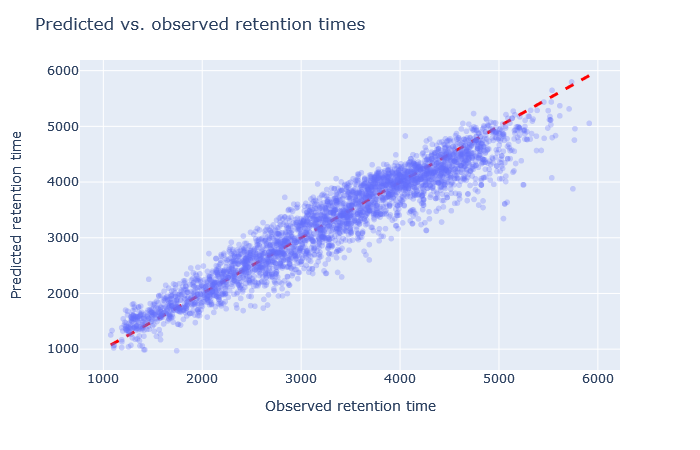
|
(results report) |
| full_hc_hela_lumos_1h_psms_aligned | 2927 (+6.8%) | 3% |
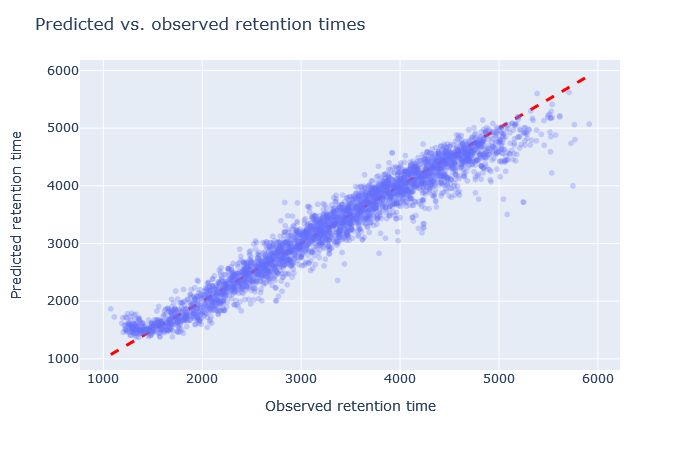
|
(results report) |
| full_hc_mod_fixed_mods | 2888 (+5.4%) | 4% |
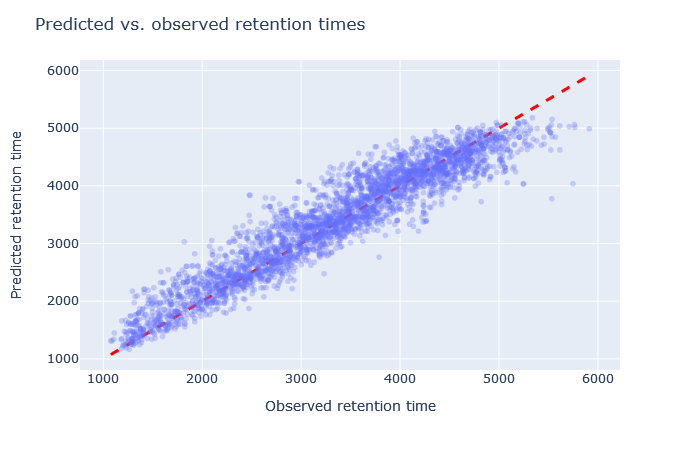
|
(results report) |
| full_hc_plasma_lumos_2h_psms_aligned | 2874 (+4.9%) | 4% |
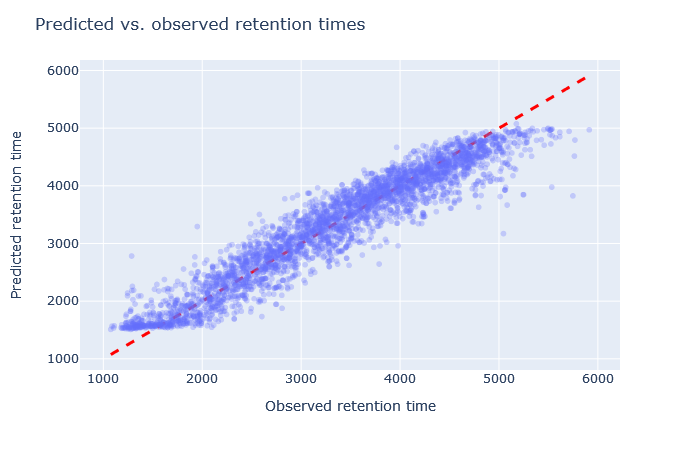
|
(results report) |
| full_hc_PXD005573_mcp | 2851 (+4.1%) | 3.5% |
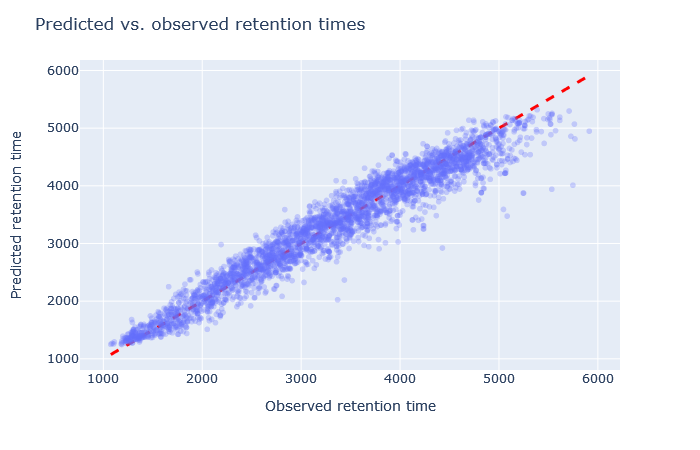
|
(results report) |
| full_hc_prosit_ptm_2020 | 2827 (+3.2%) | 5% |
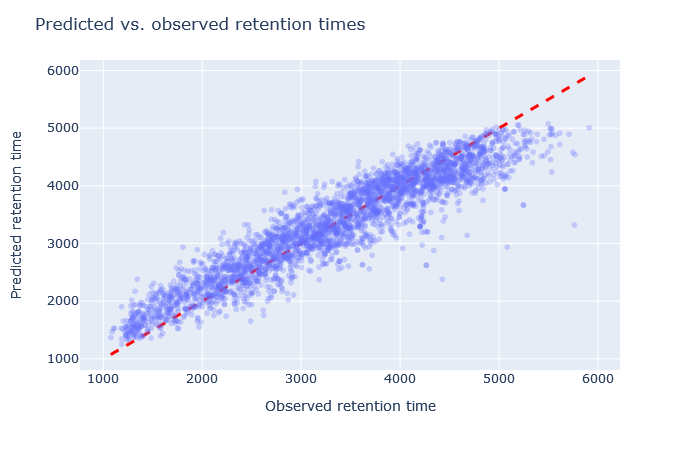
|
(results report) |
| full_hc_plasma_lumos_1h_psms_aligned | 2805 (+2.4%) | 5% |
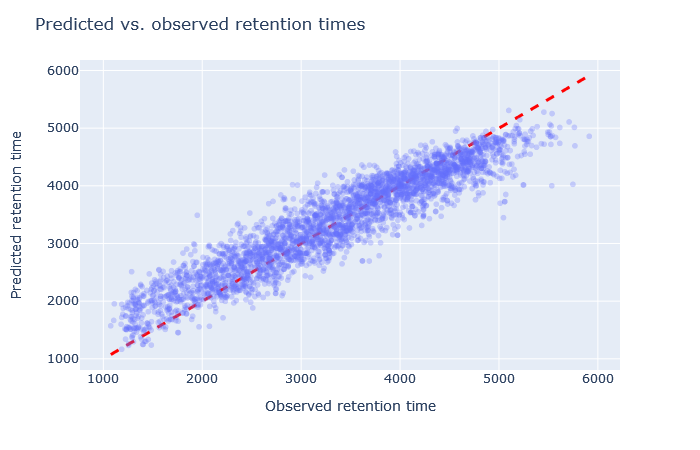
|
(results report) |
| full_hc_yeast_120min_psms_aligned | 2778 (+1.4%) | 4% |
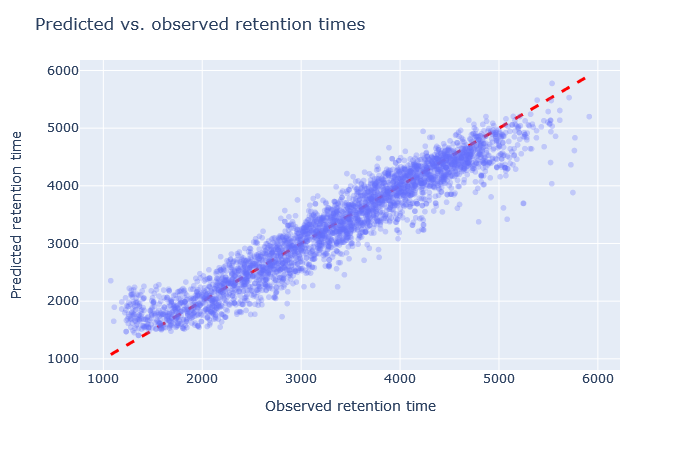
|
(results report) |
| full_hc_ATLANTIS_SILICA_fixed_mods | 2777 (+1.4%) | 14% |
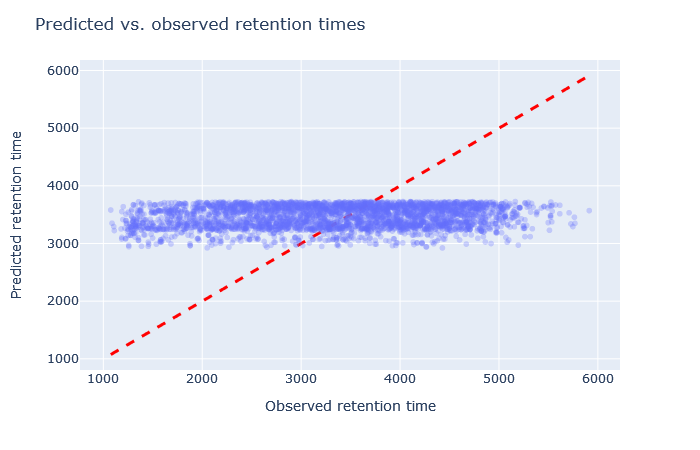
|
(results report) |
| full_hc_pancreas_psms_aligned | 2772 (+1.2%) | 5.5% |
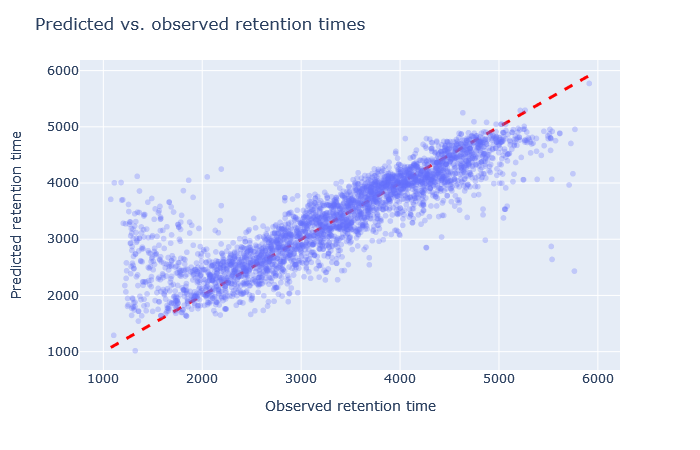
|
(results report) |
| full_hc_arabidopsis_psms_aligned | 2767 (+0.9%) | 10% |
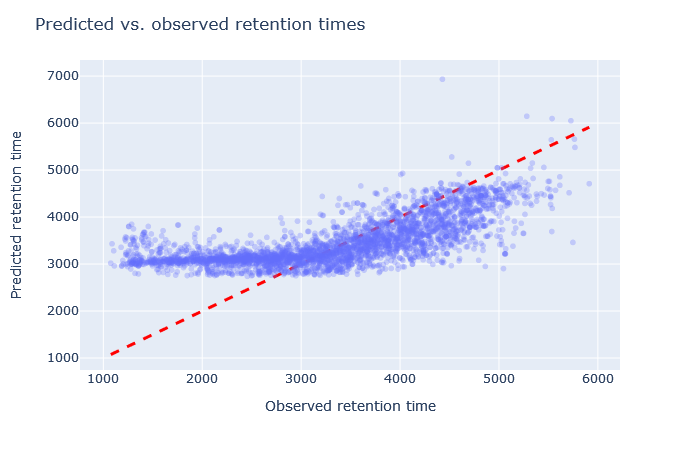
|
(results report) |
| full_hc_PXD008783_median_calibrate | 2746 (+0.2%) | 5% |
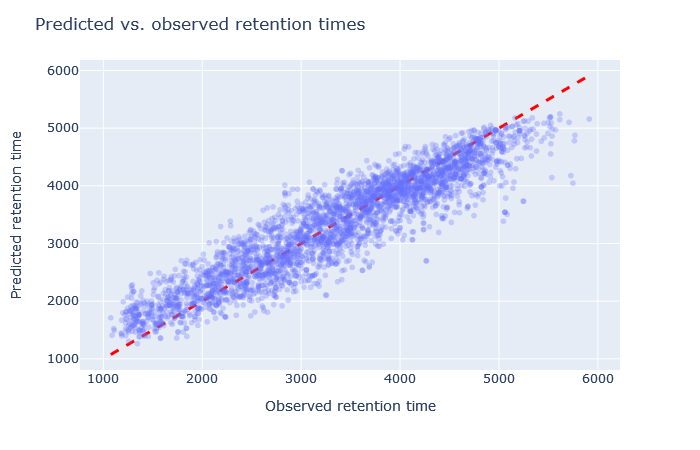
|
(results report) |
| full_hc_yeast_60min_psms_aligned | 2723 (-0.7%) | 5% |
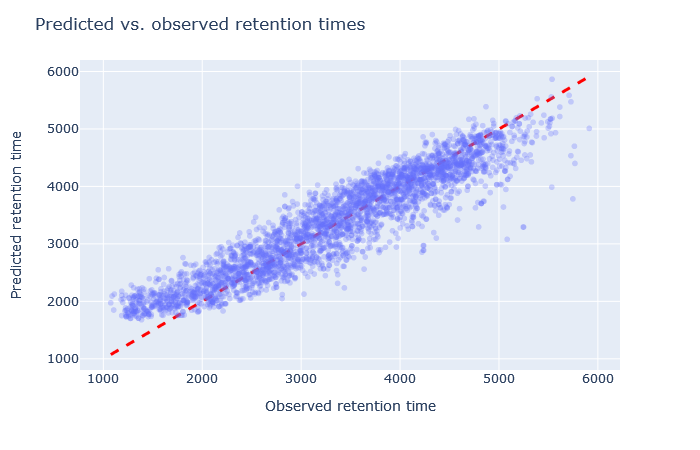
|
(results report) |
| full_hc_SCX_fixed_mods | 2693 (-1.8%) | 13% |
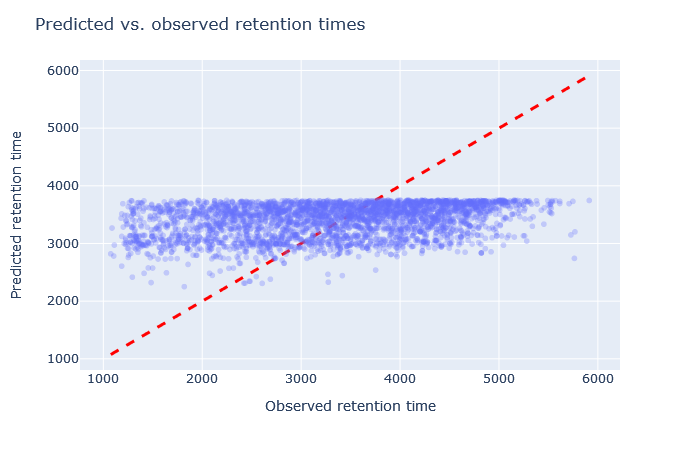
|
(results report) |
| full_hc_Xbridge_fixed_mods | 2678 (-2.3%) | 13% |
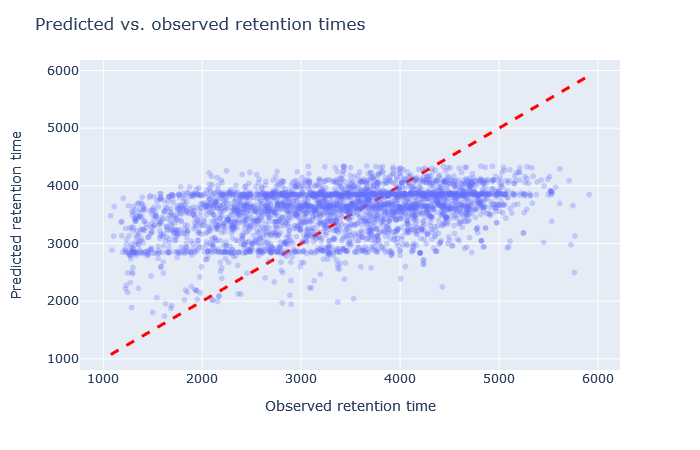
|
(results report) |
デフォルトモデルであるfull_hc_PXD005573_mcpはかなり良いモデルである事がわかります。このモデルはヒトサンプルの2時間勾配のデータでトレーニングされていますが、CPTACの酵母データセットでは勾配の時間が135分でした。修飾の設定は固定または可変ともに同じです。散布図は密集しており、RMAEはわずか3%です。このように、デフォルトモデルはユニーク配列のカウント数としては最高ではなかったものの、出発点としては妥当であると言えます。
ユニークなペプチド配列の同定数という観点では、4時間の勾配でトレーニングされたfull_hc_unmod_fixed_modsが最良でした。full_hc_PXD005573_mcpと比較すると散布図とRMAEの差はそれほど大きくなく、トレーニングデータには(未知の)何らかの要因が存在する事で、このモデルをCPTACの酵母データに対する優れた予測器にしているのだと考えられます。
選択されたモデルが間違っている事がはっきりとわかる場合もあります。例えば、full_hc_plasma_lumos_2h_psms_alignedはRMAEが4%ですが、溶出の開始時と終了時の予測があまり良くなく、このデータセットに適したモデルではありません。適合度の悪いモデルについては、散布図の形状とRMAEの両方を確認することが重要です。どちらか一方だけでは十分な評価基準とは言えません。散布図がホッケーのスティックのような形、またはそれより悪い形をしている場合は、以降の検討対象から除外します。
Keywords: DeepLC, machine learning, Percolator, retention time