|
Global ion suppression limits the potential of mass spectrometry based phosphoproteomics
Roland Felix Dreier, Erik Ahrne, Petr Broz, and Alexander Schmidt
J. Proteome Res., Article ASAP, published online November 2, 2018
The authors evaluated the impact of phosphorylation on peptide identification by LC-MS. They used an efficient dephosphorylation protocol to directly compare identified phosphopeptides with their unmodified counterparts. The samples in this study were generated in large-scale phosphoproteomics studies including frequent missed cleavages, large abundance differences and common contaminations, as opposed to the standard phosphopeptide mixtures sometimes employed.
A mix of two potent phosphatases (lambda- and alkalinephosphatase) was applied to TiO2-enriched samples from whole HeLa S3 cell lysates. The phosphatase mix removed virtually all phosphorylations after only 1 hour of treatment (>99.7% complete). When looking at the LC-MS chromatograms, the authors noticed an approximate 10-fold increase in precursor ion intensities after phosphatase treatment. Using the standard phosphoproteomics approach, they quantified 6,607 phosphopeptides from the enriched samples. When implementing the additional phosphatase treatment, precursor ion intensities were strongly enhanced and the number of quantified peptides increased by a factor of 2.3 to 15,494 peptides.
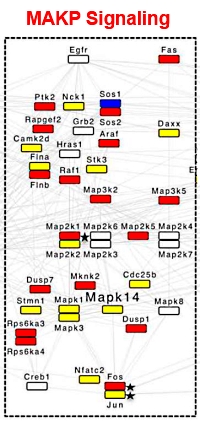
The immune signaling pathways engaged during Salmonella infection of macrophages were investigated and it was determined that the use of this phosphatase treatment doubled the number of proteins in the protein-protein interaction analysis. Sample matrix related ion suppression is the main driver of the reduced MS response/identification rates observed. This led to MS sequencing of additional peptide precursor ions, which were almost completely suppressed in the original phosphopeptide enriched sample.
|