|
プレゼンテーション2:Middle-Down検索
このプレゼンテーションでは、Middle-downの手法を利用するとProteoform(修飾位置の相違)の同定が容易になるいう研究報告を検証した際の具体的な操作内容と結果についてご説明しています。
Lys-C(KのC末端側切断、P存在未切断)やAsp-N(DのN末端側切断)のような消化酵素は、Trypsin(KとRのC末端側切断、P存在未切断)に比べて、より大きなペプチドの生成割合が高くなるように作用しますので、ピークリストの作成に注意し、適切な配列データベースを選択することにより、実験サンプル中に含まれるタンパク質の推定やProteoform(修飾位置の相違)検出の精度を高めることができます。これはMiddle-downの手法と呼ばれていますが、ちなみに、サンプル処理は一般的に Bottom-up(〜20残基) < Middle-down(20残基〜) < Top-down(Intactなタンパク質) の順に難しくなると言われています。
大きなペプチドイオンはより大きな電荷を持つ傾向がありますので、従ってプロダクトイオンも多価になる可能性が高くなります。Mascotは3価以上のプロダクトイオンであっても1価または2価と仮定して検索しますので、ピークリストを作成する際はプロダクトイオンの電荷を MH+ に変換(de-charge)し、ついでに「Instrument」設定も1価のプロダクトイオンだけを検索するように定義すると検索操作と検索結果の解釈が楽になります。
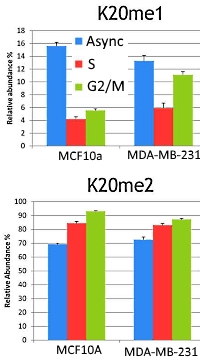
使用したMiddle-downの質量データセットは、2つの細胞株(前癌細胞株および乳癌細胞株)のヒストンH4由来で、block and release法を通じて得た非同期(ほぼG1期)、 S期、G2/M期に対応しています。
2つの細胞株で229個のproteformを同定し、そのうち非同期(G1期)由来は127個、S期由来は178個、G2/M期由来は169個でした。
プレゼンテーションに使ったスライドに詳しい説明を記載しましたので ここ をクリックしてご覧ください。
|